
In this session, we will discuss how to approach an unconscious patient. A comatose patient is always a challenge in the emergency department. The aetiology may vary from an easily correctable metabolic problem like hypoglycemia to a life-threatening one like non-convulsive epilepsy. The patient won’t be able to give history, and the clinical examination is limited as the patient is unconscious. We should know our limitations and what findings to look for in the clinical examination. It will help save time and come to an early diagnosis which can be life-saving. At the end of this session, we will be able to
1. Distinguish coma and its mimics.
2. Do focused history taking and clinical examination of an unconscious patient?
3. Identify various classic clinical findings in coma and
4. Localise coma from these findings.
Consciousness is a state of normal brain activity where one is aware of both the self and the environment and can respond to internal and external stimuli. Sleep is a standard variant of consciousness from which a person can be aroused to wakefulness. The level of consciousness is a spectrum which can vary from drowsiness through stupor to coma. In drowsiness, the patient is easily arousable and can maintain attention for some time but then goes back to sleep. In a stupor, the patient responds to painful stimuli only, while in a coma, there is no response to any external stimuli. The drowsy and stuporous patients are often confused.
Some conditions can mimic coma. These include
Locked-in state
The patient is completely paralysed and mute due to an extensive ventral pontine infarct. The patient is conscious and alert but cannot move or speak because of weakness. They can communicate through eye blink.
Akinetic mutism
The patient is partially or fully awake but is immobile and mute. They may be able to recall the events later. It occurs with lesions involving the diencephalo-mesencephalic reticular formation, frontal lobes, or severe hydrocephalus.
Abulia
It is a severe apathy caused by a lesion involving bilateral medial frontal lobes. It is a milder form of akinetic mutism. The patient has extreme mental and physical slowness and diminished ability to initiate activity. The consciousness is preserved.
Catatonia
It occurs in Schizophrenia and major depression. The patient is immobile, mute and often has waxy flexibility or catalepsy. Careful examination will show responsiveness like an eye blink to a visual threat. Though it resembles akinetic mutism, the clinical evidence of structural brain damage like hypertonia, hyperreflexia and frontal release signs are absent in catatonia. EEG will be normal in catatonia but show slowing in akinetic mutism.
Vegetative state
It occurs during recovery from a coma. The eyelids open periodically, giving a feeling of wakefulness, but the patient does not respond to internal or external stimuli. Sleep-wake cycles are present. Respiratory, cardiac and autonomic functions are present, and they will have a normal cough, yawning and swallowing.
Minimally conscious state
It is less severe than the vegetative state, where the patient will show some crude motor and vocal response spontaneously or occasionally to touch or other stimuli. The chances of recovery are better than in a vegetative state. It is always better to give a short description of the patient’s findings than use terms like vegetative or minimally conscious state. Using these terms can often produce confusion as different authors define them differently.
The anatomic substrate of consciousness
The consciousness is maintained by the interaction between the ascending reticular activating system(ARAS) and the cerebral hemispheres. The ARAS lies in the paramedian tegmental region of the pons and midbrain. It is a polysynaptic fibre system extending from the superior half of pons to the posterior portion of the hypothalamus and thalamic reticular formation. The thalamus is connected to the cortex through the thalamocortical projections. Structural and functional lesions of bilateral diffuse cortical, thalamic, hypothalamic, and brainstem tegmentum can cause coma. Metabolic causes like hypoglycemia, anoxia, and liver disease are more common causes of coma than structural lesions. Sedative drugs act at least partly by their action on ARAS. Unilateral cortical lesions only rarely produce coma. The medial longitudinal fasciculus lies between the neurons of the midbrain and pontine portions of ARAS. So coma due to brainstem lesions will affect MLF, causing eye movement abnormalities, which can often help localise coma.
Approaching a patient with unconsciousness
Initial assessment.
Whenever a comatose patient comes to the emergency room, the priority is to check if the airway, breathing and circulation (ABC) are normal. A rapid initial assessment is done, and critical interventions are done as required. Ensure that the vitals are stable, blood sugar is normal, and the patient is not having seizures. Look for subtle findings of seizures like nystagmoid jerks of the eyes. If there is any suspicion of seizures, anti-epileptic drugs should be started.
History
Once the patient is stabilised, it is crucial to get a detailed history. Getting history from a witness to the onset of coma is critical, either directly or via phone. The beginning of unconsciousness, sudden, rapid or gradual, gives a clue to the diagnosis. Sudden onset suggests trauma, seizures or SAH. Rapid onset suggests a metabolic cause like hypoglycemia, and gradual onset may indicate a subdural hematoma. Associated symptoms give diagnostic clues. Fever and vomiting at onset suggest meningitis. A history of headaches may suggest ICSOL or SAH. Past history and medication history will provide hints to diagnosis. Past h/o liver disease suggests possible hepatic encephalopathy. A recent journey to an endemic may be the only clue to cerebral malaria. History of psychiatric treatment may be the clue to deliberate self-harm. An elderly on OHA will give a hint about hypoglycemia. The history of alcoholism can be misleading. These patients are prone to SDH, hepatic encephalopathy, seizures, and nutritional and metabolic causes of coma. We must consider all these possibilities before attributing the unresponsiveness to alcohol intoxication.
General examination
A detailed head-to-foot examination is critical in the evaluation of an unconscious patient. The nutritional status, jaundice, abnormal odour, neck stiffness and fundus examination will give valuable information to diagnose the aetiology. Bruising on the skin behind the pinna called the Battle sign may be the clue to a skull fracture. An eschar on the skin may be the only clue for scrub typhus encephalitis. Multiple injection markings on the skin may be a clue to iv drug abuse producing the encephalopathy. Neck stiffness may be the clue to SAH. Fundus papilledema may be a clue for an intracranial space-occupying lesion.
Vitals gives diagnostic clues. High blood pressure may indicate hypertensive encephalopathy or PRES. A low BP may be the clue to Addisonian crisis, sepsis, MI or barbiturate poisoning.
Respiratory pattern
Respiratory patterns have localising significance in an unconscious patient. The pons has the pneumotaxic and apneustic centres, while the medulla has the inspiratory and expiratory centres. The pneumotaxic centre is located in the upper part of the pons. It controls the rate and pattern of breathing. It acts as an inspiratory off switch and is responsible for limiting inspiration. It works opposite the apneustic centre, which promotes inspiration by stimulating the medullary inspiratory neurons. The dorsal respiratory group in the medulla initiates inspiration, while the ventral respiratory group initiates expiration. It is important to remember that drugs can affect these regions and mimic a structural lesion. In normal healthy individuals, after hyperventilation with five deep breaths and CO2 wash out, there will be a brief period of apnea lasting less than 10 seconds. The stimulus for rebreathing after a pCO2 fall originates in forebrain structures. Post hyperventilation apnea lasts for 20-30 seconds in patients with bilateral hemispheric lesions.
Cheyne -stokes breathing.
There are brief periods of hyperpnoea regularly alternating with an even shorter duration of apnoea. After the apneic phase, respiratory movement’s amplitude increases to a peak before slowly waning off to the following apnea. It represents a more severe degree of normal post-hyperventilation apnea due to bilateral cortical dysfunction. The respiratory drive becomes dependent on pCO2 levels as the smoothening effect of forebrain structures is lost. In the hyperventilation stage, the patient will be more alert. The pupils will become more dilated from the otherwise diencephalic miotic pupil, and decorticate posturing may disappear. Cheyne-stokes breathing is caused by widespread cortical dysfunction, involvement of descending pathways up to upper pons and metabolic disturbances like uremia, cardiac failure, diffuse hypoxia etc. New-onset Cheyne-Stokes breathing may suggest an impending transtentorial herniation in a patient with a supratentorial mass lesion.
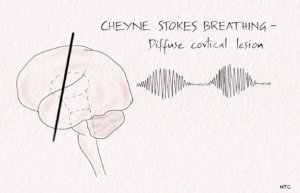
Fig 1 – Cheyne Stokes breathing
Brainstem hyperventilation
Patients with midbrain and pontine lesions can have rapid and prolonged hyperventilation. Lowering the local CSF pH and stimulating the respiratory centre of the medulla is one of the postulated mechanisms. Kussmaul’s breathing is deep, rapid breathing seen in metabolic acidosis.
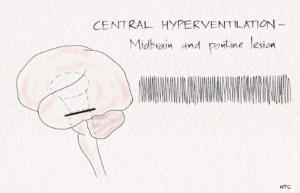
Fig 2- Central hyperventilation
Apneustic breathing
In apneustic breathing, there is a prolonged inspiratory time followed by a long inspiratory pause. The air is retained for several seconds and released. The expiratory time is short, and a pontine lesion usually causes it.
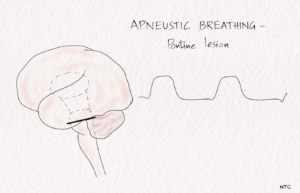
Fig-3 Apneustic breathing
Cluster breathing
It is a cluster of breaths following each other in an irregular sequence. It results from low pontine or high medullary lesions.
Ataxic breathing
The breathing pattern is entirely irregular. It is called the atrial fibrillation of breathing. The inspiration is of different amplitudes and lengths and intermixed with variable periods of apnea. It is often seen in terminally ill patients and suggests impending respiratory failure. The site of the lesion is the dorsomedial medulla. A paramedian medullary infarct, bleed, or tumour can produce it. The classical breathing described by Biot in severe meningitis is ataxic.
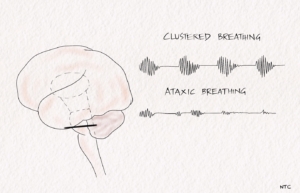
Fig 4 -Ataxic and cluster breathing in low pontine and medullary lesion.
Central hypoventilation
The pathway of voluntary respiration from the cortex descends via the corticospinal tract. The automatic respiration fibres from the medulla descend in the ventrolateral cord, with anatomic separation of inspiratory and expiratory pathways. Central hypoventilation can be caused by unilateral caudal brainstem infarction. In the Ondine’s curse, there is a loss of automatic breathing in sleep in non-comatose patients. The involvement of pontomedullary reticular formation and nucleus ambiguus can cause loss of automatic respiration or Ondine’s curse. If the nucleus tractus solitarius is also involved, severe respiratory failure with both voluntary and automatic responses is lost. Bilateral high cervical cord lesion can also produce central hypoventilation.
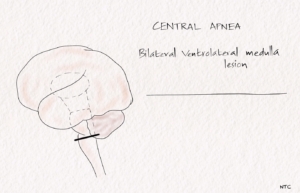
FIg-5 Central apnoea
Neurological examination
The neurological examination is grossly limited in an unconscious patient. There are only a few findings one can elicit in these patients. These include
1. Level of consciousness or GCS
2. Pupil
3. Eye movements
4. Brain stem reflex
5. Motor response
Level of consciousness
The neurological examination starts with defining the state of consciousness using the Glasgow coma scale. GCS is simple, can be done by any health care worker and is easily reproducible. It objectively assesses how the patient progresses and indicates the need to reassess or change management. It should be documented at arrival and periodically after that.
Pupil
The pupillary light reflex is resistant to metabolic dysfunction. Pupillary abnormalities, especially when unilateral, indicate structural lesions of the midbrain or third cranial nerve. The size, shape, symmetry, and response to light of the pupil give important diagnostic clues. It is critical to rule out any medication given to the patient that might affect the pupillary size and reactivity. Atropine, glutethimide, barbiturates, succinylcholine, lidocaine, phenothiazines, methanol and aminoglycoside antibiotics can cause an unreactive pupil. Sedative drugs affect pupils in very high doses only. Hypothermia and anoxia can also cause an unreactive pupil, which, if persistent, carries a poor prognosis.
Diencephalic pupil
Bilateral diencephalic dysfunction, metabolic coma, and sleep can cause small pupils that react well to light.
Unilateral hypothalamic lesion
A unilateral hypothalamic lesion can cause miosis and anhidrosis on the side of the body, ipsilateral to the lesion.
Midbrain lesions
Tectal or pretectal lesions affecting the posterior commissure abolish the light reflex. The pupils are mid-sized or slightly large. The pupil can have spontaneous oscillations in size called the hippus. Ciliospinal reflex, the dilatation of the pupil on pinching the skin of the neck, is present.
Tegmental lesions involve the third nerve nucleus. There is irregular constriction of the sphincter of the iris. This results in a pear-shaped pupil or displacement of the pupil to one side called the midbrain corectopia. The pupil is mid-sized and lacks light or ciliospinal reflex.
Pontine tegmental lesion
The pontine tegmental lesion will cause a small pupil due to the interruption of descending sympathetic pathways. Pontine haemorrhage will cause a pinpoint pupil. It is due to sympathetic damage and parasympathetic irritation. The pupil will react when observed under a magnifying glass.
Lateral pontine, lateral medulla and ventrolateral spinal cord lesions
Lesions in these sites produce an ipsilateral Horner’s syndrome.
Oculomotor nerve palsy
Uncal herniation can cause ipsilateral oculomotor palsy. Pupillary dilatation occurs before extraocular muscle involvement.
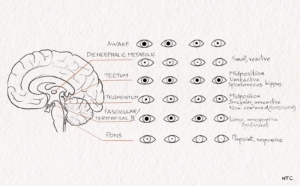
Fig -6 Pupils in darkness and light in various brain lesions
Eye movements
Voluntary eye movements cannot be tested in an unconscious patient. The examination of ocular motility depends on the reflex eye movements, namely oculocephalic reflex and oculovestibular reflex. Oculocephalic reflex is tested by dolls eye manoeuvre. It is elicited by moving the head from side to side through about 70 degrees or vertically from passive neck flexion to passive neck extension. Eye movements in the direction opposite to the head movement are noticed. It depends on the integrity of the ocular motor nuclei and their interconnecting tracts that extend from the midbrain to the pons and medulla. The movement is usually suppressed in a conscious patient with an intact frontal lobe. So the presence of oculocephalic reflex suggests reduced cortical influence and intact brainstem pathways. It is lost in extensive brainstem damage and deep metabolic coma. It should not be tested in patients with a suspected spine injury.
The oculovestibular reflex is elicited by instilling cold or warm water into the external auditory canal. Before doing the test, it is essential to make sure that the tympanic membrane is intact. 20 ml of ice-cold water is instilled into the external auditory canal over 30 seconds after keeping the head elevated by 30 degrees. After a brief latent period, both eyes will deviate to the side of cold water irrigation. In the unconscious patient, the central corrective nystagmus in the opposite direction will not occur. The COWS acronym of oculovestibular reflex is in the direction of nystagmus, which is in the direction of the central fast component. This will not happen in an unconscious patient. The oculovestibular reflex is resistant to metabolic changes and is one of the last brain stem reflexes to disappear in a deep metabolic coma. It is lost in structural lesions of the brainstem.
A lot of spontaneous eye movement abnormalities can be seen in a comatose patient. Many of them have localising value. Standing near the patient and observing the eyes for a minute or two will help pick up most spontaneous eye movement abnormalities.
Periodic alternating gaze or ping pong gaze
There will be roving eye movement from one extreme of the horizontal gaze to the other and back. The horizontal rowing eye movements suggest an intact brainstem. It usually occurs in bilateral cerebral dysfunction.
Repetitive divergence
It is occasionally seen in patients with metabolic encephalopathy. The eyes are mid-positioned on slightly divergent at rest. They slowly deviate out to the extreme and rapidly return to the primary position.
Nystagmoid jerk of one eye
It can be vertical, horizontal or rotatory. It usually suggests a mid or lower pontine lesion. Patients with non-convulsive electrographic status epilepticus can have horizontal or vertical nystagmus jerks, which may be the only clue to the seizure.
Ocular bobbing
There is brisk, bilateral downward movement of the eyes with a slow return to mid-position. It occurs with pontine lesions. Ocular dipping is slow downward eye movement with a fast return to mid-position. It occurs in diffuse brain dysfunction. A way to remember these is that ocular dipping is like dipping hands in hot water. The hand goes down slowly. On touching the hot water, it comes up quickly. Ocular bobbing is the reverse, with fast movement downwards and slow upward.
Reverse ocular bobbing and dipping
They are similar to ocular bobbing and dipping, but the direction of eye movement is upwards rather than downwards. In reverse ocular bobbing, there is fast upward eye movement followed by a slow return to mid position. In reverse ocular dipping, there is slow upward eye movement followed by a quick return to mid-position.
Pretectal pseudo bobbing
There are arrhythmic, repetitive downward and inward eye movements in a V pattern. The frequency range from one every three seconds to two per second. It is usually seen in acute hydrocephalus and usually warrants emergency surgical intervention.
Vertical ocular myoclonus
It is a pendular, vertical isolated eye movement seen in large pontine strokes. The frequency is about 2Hz. It may be associated with palatal myoclonus.
Abnormality of gaze
Lateral gaze
There are two lateral gaze centres. The frontal gaze centre will help you look to the opposite side, and the pontine gaze centre or para-pontine reticular formation(PPRF) to the same side. So when there is a right frontal lesion, the patient will have a gaze palsy to the left and gaze preference to the right. One easy way to remember this is the patient’s eyes will deviate to the side of frontal lobe damage. Remember that a left pontine lesion can also cause gaze preference to the right. In a left pontine lesion, the hemiplegia will be on the right, while in a right frontal lesion, the hemiplegia will be on the left. The oculocephalic and oculovestibular manoeuvres can move the eye to the paretic side in a frontal lesion but not in a pontine lesion.
Disconjugate gaze
The involvement of the Medial longitudinal fascicle (MLF) will result in ipsilateral adduction impairment with normal vertical eye movements and pupils. The abducting nystagmus on the opposite side in inter-nuclear ophthalmoplegia(INO) will not be present in a comatose patient. MLF involvement is commonly bilateral in an unconscious patient.
Vertical gaze
In a patient with a light coma, an upward gaze can be tested by gently touching the cornea with a wisp of cotton after keeping the eyelids open. The eyeball tends to role upwards due to the Bell’s phenomenon. The vertical eye movements in a comatose patient can also be tested with doll’s eye manoeuvre and vestibulo-ocular reflex. Irrigating both ears with warm water induces deviation upward, and cold water causes downward movement.
Disconjugate vertical gaze in the resting position is called skew deviation. Persistent eye deviation below the horizontal median usually suggests a structural lesion of the midbrain tectum. Thalamic haemorrhage can cause a tonic downward deviation of the eyes with convergence. Tonic upgaze can occur after severe hypoxia. Midbrain lesions can produce vertical gaze palsy.
Motor system examination
Examining the tone and reflexes has less clear localising value in an unconscious patient. In most comatose patients, both plantar are usually upgoing. However, all attempts should be made to find out any localising signs. If present, it helps in identifying the aetiology of coma. Deep sternal pain can unmask the facial deviation or demonstrate the lack of movements on one side in a stuporous patient. Metabolic coma, except for hypoglycemia, usually has no localising signs.
Decorticate rigidity is characterised by adduction of the shoulder and arm, flexion at the elbow and flexion and pronation at the wrist. The leg is extended at the hip and the knee. It can occur in metabolic encephalopathy or structural lesions involving the cerebral hemispheres.
Decerebrate rigidity is associated with extension and pronation of the upper extremity and extension of the lower extremity with forced plantar flexion. Painful stimuli can cause opisthotonus with the hyper extension of the trunk and hyper-pronation of the arms. It is associated with severe metabolic coma and structural lesions involving the brainstem. In experimental animals, decerebrate rigidity is caused by a transection at the collicular level below the red nucleus. But clinically, most of the cases are produced by deep metabolic coma.
Flaccidity in an unconscious patient, especially after a few days, should make one consider the possibility of critical illness polyneuropathy. In patients on steroids especially, the possibility of muscle necrosis or critical illness myopathy has to be considered.
That finishes this session. I was planning to finish the first season of clinical localisation in 12 sessions. But some of you had requested for localisation of bladder and cortical function. So I thought I would add those sessions as well. If you have any suggestions or comments, please feel free to message me on the Twitter account or on the neurology teaching club website. Your comments and response are my fuel to keep going. Please do give a five-star rating and write a review on the Apple podcast and Spotify. Please share the podcast with your friends and on social media platforms if you found the content useful.